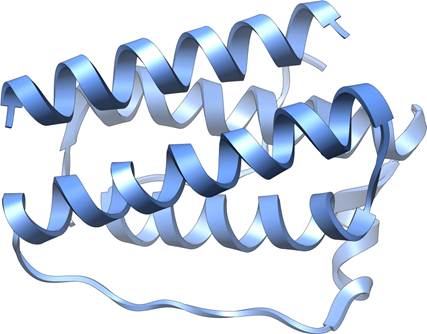
Leptin structure
In the mid-1990s, a breakthrough in obesity research fundamentally changed our understanding of how the body regulates weight, hunger, and metabolism. The discovery of leptin, a hormone that would prove central to energy balance, emerged from decades of research involving an unusual laboratory mouse strain and culminated in findings that would help patients with rare but devastating metabolic disorders. This is the story of how scientific curiosity, persistence, and a colony of obese mice led to one of the most significant discoveries in modern endocrinology.
The Mystery of the OB Mouse
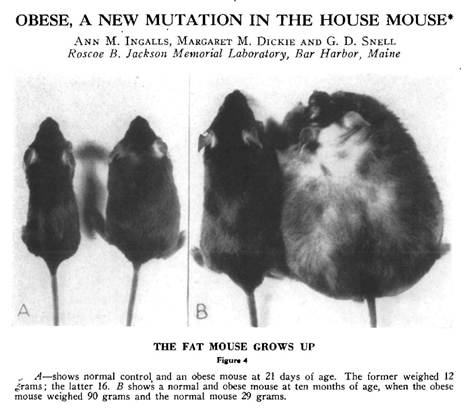
The story begins in 1949 at the Jackson Laboratory in Bar Harbor, Maine, where researchers noticed something extraordinary: a spontaneous mutation had produced mice that became massively obese. These mice, which came to be known as the “ob/ob” mice (short for “obese”), weighed nearly three times as much as their normal littermates. They were voraciously hungry, ate constantly, and developed severe obesity along with diabetes-like symptoms.
For decades, these ob/ob mice remained a biological curiosity. Scientists knew the obesity was caused by a recessive genetic mutation, meaning both copies of a particular gene had to be defective for the condition to appear, but the identity and function of that gene remained elusive. The mice became an invaluable research tool, but the fundamental question persisted: what was different about these animals at the molecular level?
Adding to the intrigue was the existence of another mutant mouse strain called “db/db” (for “diabetes”), discovered in the 1960s. These mice had a remarkably similar phenotype to the ob/ob mice—they too were obese, constantly hungry, and diabetic. Yet they had a different genetic mutation. The relationship between these two strains would prove crucial to understanding the biology of weight regulation.
Parabiosis: A Clue from Connected Mice
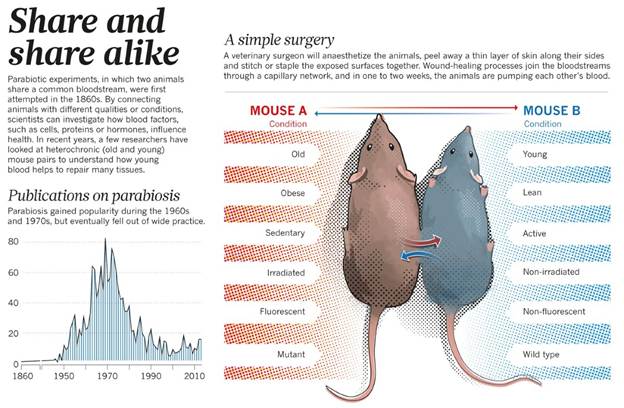
A pivotal insight came from elegant experiments conducted in the 1970s by Douglas Coleman at the Jackson Laboratory. Coleman performed parabiosis experiments—surgically joining two mice so they shared a circulatory system. When he connected an ob/ob mouse to a normal mouse, something remarkable happened: the obese mouse ate less and lost weight. However, when he connected a db/db mouse to a normal mouse, the normal mouse stopped eating and became emaciated, while the db/db mouse remained obese.
These results suggested a powerful hypothesis: the ob/ob mice lacked some circulating factor that suppressed appetite and promoted energy expenditure, while the db/db mice produced this factor but couldn’t respond to it. In other words, ob mice couldn’t make the signal, while db mice couldn’t receive it. The circulating factor was being produced by the normal mouse and affecting the ob mouse through their shared bloodstream, but the db mouse appeared to be overproducing the factor to no effect, overwhelming its normal partner.
Coleman’s work provided a conceptual framework, but identifying the actual molecule would require tools that didn’t yet exist. The answer would have to wait for the molecular biology revolution.
Jeffrey Friedman and the Hunt for the OB Gene
Enter Dr. Jeffrey Friedman, a physician-scientist at Rockefeller University who became fascinated with understanding the genetic basis of obesity. In the late 1980s, Friedman set out to identify and clone the ob gene using an approach called positional cloning, a painstaking technique that involves tracking down a gene based solely on its chromosomal location without any prior knowledge of what the gene does or what protein it produces.
This was an audacious undertaking. The mouse genome had not been sequenced. The techniques for positional cloning were still being developed. The project would require mapping the mutation to a specific chromosomal region, narrowing down that region through genetic linkage analysis, identifying candidate genes, and then proving which one was responsible for the obesity phenotype.
Friedman’s laboratory spent years on this pursuit. Using genetic markers and analyzing thousands of mice from breeding crosses between ob/ob mice and normal mice, they progressively narrowed down the location of the ob gene on mouse chromosome 6. They eventually identified a region containing only a handful of genes. The team then systematically examined each candidate gene, looking for differences between normal mice and ob/ob mice.
The breakthrough came in 1994. After nearly eight years of intensive research, Friedman’s team identified the ob gene. The gene coded for a previously unknown protein that was expressed primarily in white adipose tissue—body fat. In ob/ob mice, the gene contained a mutation that prevented production of functional protein. The team named the protein “leptin,” derived from the Greek word “leptos,” meaning thin.
The Science Behind the Discovery
The identification of leptin represented a triumph of molecular biology techniques. Friedman’s team used several key approaches:
Genetic Mapping: By breeding ob/ob mice with normal mice from different strains, researchers could track which genetic markers co-segregated with the obesity phenotype across generations. This allowed them to progressively narrow the chromosomal region containing the ob gene from millions of base pairs down to a manageable segment.
Physical Mapping: Once the region was identified, the team created overlapping clones of DNA fragments covering that area—a technique called “chromosome walking.” This allowed them to systematically examine the DNA sequence of the candidate region.
Candidate Gene Analysis: Within the narrowed region, researchers looked for sequences that resembled known genes. They identified several candidates and examined their expression patterns and sequence variations between normal and obese mice.
Mutation Analysis: The crucial evidence came when they found that ob/ob mice had a mutation in one particular gene that caused a premature stop signal in the protein-coding sequence. This meant the gene couldn’t produce a functional protein. A different natural mutation in another strain of obese mice affected the same gene differently but with the same result—no functional leptin.
Expression Studies: The researchers demonstrated that the ob gene was expressed primarily in adipocytes (fat cells) and that the level of expression correlated with the amount of body fat. They showed that normal mice produced leptin in proportion to their fat stores, while ob/ob mice produced none.
The final proof came from administering leptin to ob/ob mice. When given injections of the leptin protein, these previously insatiable, obese mice ate less, became more active, and lost weight dramatically. Their metabolism normalized. It was a stunning validation of decades of hypothesis and years of painstaking molecular work.
What Does Leptin Do?
The discovery of leptin revealed a sophisticated system for regulating body weight and energy balance. Leptin functions as an adiposity signal, essentially, fat tissue uses leptin to communicate with the brain about the body’s energy stores.
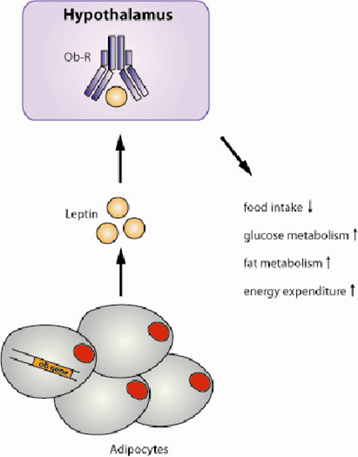
Here’s how the system works: adipocytes produce leptin in proportion to the amount of fat they contain. More body fat means more leptin production. The leptin circulates in the bloodstream and travels to the brain, where it binds to receptors concentrated in the hypothalamus, a region critical for regulating appetite, metabolism, and energy expenditure.
When leptin levels are adequate, the brain receives the signal that energy stores are sufficient. This triggers several responses: appetite decreases, energy expenditure increases through mechanisms like thermogenesis (heat production), and the body shifts toward using stored energy. The hypothalamus also regulates other hormones and physiological processes to maintain energy balance.
Conversely, when leptin levels drop—as happens during weight loss or starvation—the brain interprets this as a signal that energy stores are depleted. Hunger increases, metabolism slows to conserve energy, and the body shifts toward storing any available calories as fat. This is why losing weight becomes progressively harder—the body fights to defend its fat stores through leptin-mediated mechanisms.
The db/db mice, it turned out, had mutations in the leptin receptor gene. They produced leptin, often at very high levels, but their brains couldn’t respond to it. This explained Coleman’s parabiosis results: the circulating leptin from db mice entered the normal mice and suppressed their appetite, while the db mice themselves remained obese because they couldn’t sense the hormone.
From Mice to Humans: The Leptin Connection
Friedman’s discovery immediately raised a critical question: was leptin relevant to human obesity? The answer proved complex. In 1997, researchers identified the first humans with congenital leptin deficiency, children from consanguineous families who, like ob/ob mice, had mutations in both copies of their leptin gene.
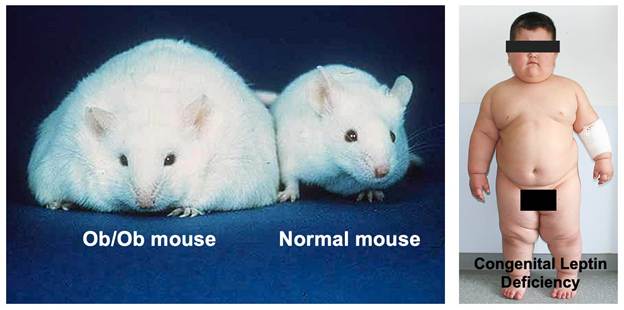
These children presented with a dramatic phenotype: they were born at normal weight but rapidly developed severe obesity in early childhood, often reaching extreme weights by age 5 or 6. They had insatiable hunger, constantly seeking food, and displayed food-seeking behaviors that disrupted family life. They also had delayed puberty due to hypogonadotropic hypogonadism, leptin’s absence prevented normal sexual maturation.
The medical implications extended beyond obesity. Many patients with congenital leptin deficiency developed severe metabolic complications:
Lipodystrophy-like Features: While the genetic form involves too little leptin from too much fat, patients with lipodystrophy, a group of disorders characterized by loss of body fat, also have leptin deficiency, but from the opposite cause: too little fat tissue. Whether from genetic mutations or acquired fat loss, the lack of leptin causes similar metabolic derangements.
Severe Hypertriglyceridemia: Leptin deficiency causes marked elevations in triglycerides, sometimes reaching levels over 1000 mg/dL or higher. This creates visible lipemia—the blood plasma appears milky white due to excess fat particles. Such extreme elevations dramatically increase the risk of pancreatitis, a potentially life-threatening inflammation of the pancreas.
Fatty Liver Disease: Without adequate leptin signaling, the liver accumulates excessive fat, leading to hepatic steatosis and potentially progressing to more severe liver damage.
Insulin Resistance and Diabetes: The metabolic dysfunction extends to glucose metabolism, with many leptin-deficient patients developing insulin resistance and type 2 diabetes at young ages.
Immune Dysfunction: Leptin plays a role in immune function, and deficient patients often have increased susceptibility to infections.
Leptin Replacement: A Remarkable Treatment
When leptin-deficient patients were identified, the obvious treatment approach was leptin replacement therapy—essentially, providing the hormone these patients couldn’t make. The results were nothing short of miraculous.
Children and adults with congenital leptin deficiency who received recombinant leptin (metreleptin) experienced dramatic transformations. Within weeks to months of starting treatment, patients showed:
Marked Weight Loss: Patients lost substantial weight, sometimes 50% or more of their body weight over the first year of treatment. The weight loss continued until they reached near-normal weight ranges.
Normalized Eating Behavior: The constant hunger disappeared. Patients who had been consumed by food-seeking behavior developed normal appetite regulation. Families reported life-changing improvements in quality of life.
Resolution of Metabolic Abnormalities: Triglyceride levels plummeted from dangerous elevations to normal ranges within weeks. The lipemia resolved. Fatty liver improved. Glucose metabolism normalized.
Sexual Development: Children with delayed puberty began normal pubertal progression. Adults with hypogonadism experienced normalization of sex hormones.
Improved Quality of Life: Beyond the physical changes, patients reported profound improvements in well-being, energy, and ability to participate in normal activities.
The treatment was equally effective in patients with acquired lipodystrophy. Whether from genetic causes, autoimmune destruction of fat tissue, or HIV treatment-related fat loss, patients with severe lipodystrophy and leptin deficiency showed similar dramatic responses to leptin replacement. The FDA approved metreleptin in 2014 specifically for treating complications of leptin deficiency in patients with congenital or acquired lipodystrophy.
The Broader Impact and Unexpected Discoveries
The leptin story illustrates both the power and the limitations of translational research. While leptin replacement is spectacularly effective for the rare patients with leptin deficiency, it largely failed as a treatment for common obesity. Most obese people have high leptin levels—their problem isn’t leptin deficiency but leptin resistance, where the brain fails to respond appropriately to the hormone’s signal. This parallels type 2 diabetes, where the problem is usually insulin resistance rather than insulin deficiency.
Nevertheless, leptin research opened entire new fields of investigation. Scientists discovered that leptin is part of a complex network of hormones and neural circuits regulating energy balance. The hypothalamic pathways that respond to leptin have been mapped in extraordinary detail. We now understand that leptin regulates not just appetite and metabolism but also immunity, reproduction, bone metabolism, and many other physiological systems.
The discovery validated the concept that body weight is biologically regulated—that the body has sophisticated mechanisms for sensing and defending its energy stores. This shifted the conversation about obesity from one of simple willpower to recognition of underlying biological drives that make weight loss and maintenance challenging.
Legacy of Discovery
Jeffrey Friedman’s identification of leptin stands as one of the landmark achievements in metabolism research. The work earned him numerous awards, including the Lasker Award (often called “America’s Nobel”) and the Shaw Prize. More importantly, it provided:
Understanding of Weight Regulation: Leptin revealed that body weight isn’t simply a matter of calories in versus calories out, but involves active biological regulation through hormonal signaling between fat tissue and the brain.
Treatment for Rare Diseases: For patients with congenital leptin deficiency or lipodystrophy, leptin replacement is genuinely life-saving, preventing deadly complications like pancreatitis and providing normal quality of life.
Research Framework: The discovery established paradigms for studying energy homeostasis and identified key neural circuits in the hypothalamus that have become targets for developing new metabolic therapies.
Validation of Genetic Approaches: The success of positional cloning to identify leptin validated this approach and paved the way for identifying many other disease genes.
The story of leptin demonstrates how basic research—studying mutant mice in a laboratory—can lead to profound clinical applications. Those ob/ob mice that puzzled researchers in 1949 eventually revealed fundamental truths about how our bodies regulate weight and led to treatments that transformed lives for patients with devastating metabolic disorders.
The journey from a curious mutation in mice to a life-saving treatment for humans took nearly half a century. It required insights from genetics, molecular biology, endocrinology, and clinical medicine. It stands as a testament to the value of basic research and the unexpected ways that understanding fundamental biology can ultimately benefit human health. The ob mouse, once just an oddity in a research colony, became the key to unlocking one of metabolism’s most important secrets.